����
����
��������������£������߿ɷ������������
�߹ǹ������δ��ˡ������ѡ�Ѫ�غ�
�����������������ǻ�ڵ�ֱ�������dz�����ԭ��ͷ�����˺���ʶ
�����ߣ�����ѪҺ��θ���������������Դ�Է�����
��ˮ�������������Ҳ���ټ������������Է������Ĵ����߷�����
���Ժ���˥����Խ��Խ��ע�⣻�������Ѫ����Һ���࣬���ۺ��֬��˨�����Լ����˺��Ⱦ��������ɺ��������ۺ�������֪ԭ��
1.�ݿ� ���������ڴ���ʧѪ��ɵĵ�Ѫ��������������������ͣ�ͬʱҲ��ɷ�Ѫ�������١����ڷ�Ѫ�����ļ��ٺ�ԴԴ���ϵؽ�����ѭ����������˨�ӣ��ɶ�����Ѫ�ܴ������谭���彻���Ľ��С��ƻ���Ѫϸ������֯�ֽ���������֧���ܺͷ�СѪ����������ʹëϸѪ��ͨ�����ӣ�����μ��ʳ�Ѫ��ˮ�ף�ʹ���������Ӵ�����ڳ־����ݿ˵Ļ����ϣ������������أ��������Һ����Ѫ�ȣ����ɵ��º��������ۺ�����
2.֬��˨�� ֬��˨���Ƕ���ۺ��IJ���֢�����֬���ο�������С������ʹ֮���š�С֬���ο���ɢ�ںܶ�СѪ�ܣ���ɹ㷺��ѭ��˨����ͬʱ����֬����֬ø�������£��ֽ������֬���ᣬ����ɵĻ�ѧ�����Է�Ӧ���ɵ���
��ˮ���ͷγ�Ѫ���ٴ��ϱ����е���Ѫ֢���Ƿι�����һ����Ҫָ�ꡣ
3.��Һ���� �����ش������У�����Ӧ����Ӧ��ˮ���������ķ�Ӧʱ���Ϊ�־ã�������72h����ˣ��˺������Һ��ʹ����ˮ���������ڣ�������ϸ����Һ����ͬʱ�����������Һ����ϡ��Ѫ�����ף�����Ѫ���Ľ�����ѹ����ʹ
��ˮ�����ء����⣬������౾����ֱ���ܵ����ֲ�ͬԭ�����������ˡ��������ݿ˻�ŧ��֢�ȣ�������������������ˮ�֡���ˣ���ʹ��������Һ������Ҳ�����
��ˮ�������ԣ���Һ�����ڷ������Ժ��������ۺ�������������У���ռ���൱��Ҫ�ĵ�λ���������о�������
��ˮ��ʱ����֫��С���ͷ�ëϸѪ�ܾ�ˮѹ�IJ�𣬷�����֫ëϸѪ��ѹΪ16mmHg��С��ëϸѪ��ѹΪ15.4mmHgʱ���ŷ���ˮ�ף�����ëϸѪ��ѹΪ7.6mmHgʱ��������
��ˮ����
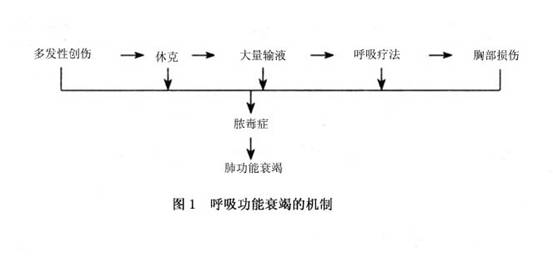
4.��Ⱦ ��ŧ�Ը�Ⱦ��ʹϸ�����ػ�ϸ��������������ѭ�������ڶ��������£������ͷų�Ѫ�ܻ������ʣ���5-�ǰ����鰷�����������Ӱ��ȣ���ʹëϸѪ��ͨ�����ӡ���Ⱦ������ת�����β����Ӷ������ι���˥�ߡ����ݿˡ���Դ��˺ʹ�����Һ�����أ�������ʹ���˷���ŧ��֢���䷢����������˥�ߵĻ��Ƽ�ͼ1��
5.�Դ��� �����Դ��˳�����
��ˮ����������Ϊ�Դ��˿��Լ���ǿ�ҵĽ����嶯������������ĩ��Ѫ���������漴Ѹ�ٷ�����������˥�ߺ�
��ˮ������Ԥ��Ӧ�æ���������������ҩ���ɷ�ֹ������������ִ��˺�
��ˮ���Ļ�Һ�ڵ����ʺ����ܸߣ��ʳ���ѹ��ˮ���⣬��������ͨ��ˮ�����صĴ��ڡ�
6.���� ������Ϊ������������ۺ�����ԭ��֮һ�������ܵ����ӡ���������������θ�������Ƿdz����ص������С��pH����2.5�����Է����Ҳ��������غ��������ѧ�Է��ͷβ���Ⱦ���Ӷ����º���˥�ߡ�
7.���ж� ����˥��ʱ�����ø�Ũ�������ƣ�������ʹ�÷�����ɷ����������ж�����Ҫ��������������ѹ��������ʱ�䣬������ѹ������ʱ�����������Ի���Ŀ������������ж�ʱ��֧���ܵ���ë�˶����ܵ��������ơ�100%������6h�����ɲ�����֢״�ļ���֧�����ס�Sevittͨ������ʬ����������Ϊ��Ĥ�������Է���Ϊ�˷����ж�������������Ҫ�IJ��������ı���ͨ��-��������ʧ��������ѪҺ�����ε�ˮ�ס����š�ͻ�����ά���������ʹ�������������������࣬�γɾ���Ѫ�������ӣ����Dz��������Եĵ���Ѫ֢����������������ɢ�ϰ���������̼�ų����裬��ʱ��ʹ�����Ũ��������������߶�������ѹ��ֻ�ܼ��ضԷεĶ�����ʵ���пɼ����ﳣ��������ȱ��������ͣ����
��������
�������ƣ�������ڱ��������Ļ��ƻ��������������һЩ����ѧ����������Ļ��ƣ���û��һ���ܽ������еķ��������ÿһ���岡�˵ķ���������������ֻ�����������
1.
��ˮ���IJ��� ���ˡ��ݿ˼������²����ص�ʹ��ѭ��ѪҺ�������㣬����ֱ�����ݺ�ëϸѪ�ܡ�ͨ���������ʣ���Ѫ˨��Ѫ�ܻ������ʻ���֢��Ӧ���ʵ��������-ëϸѪ��Ĥ����ʹ��ͨ�����ӣ�Һ���ɴ�ëϸѪ����©�����ݻ�����У�����
��ˮ�������⣬�ݿˡ����˻������²�����ʹ��Ѫ����ע���㣬�Դ�л���ý��ͣ�����������
��Ѫ�ܾ������Ӷ�����ξ���ѹ���ӡ���Һ������Ҳ�������
��ˮ���IJ�����
���˹۲쵽Ѫѭ���е���ϸ����ѪС�弰��֯����ϸ���У������и�����֢���ʣ�����ϸ������ø��ˮ��ø����֬ø�������DZ��ͷŽ����ѭ���У����ʹ����ëϸѪ��Ĥ�����㷺����ʹͨ�����ӣ������ʡ�Ѫϸ����Һ����©��Ѫ���⡣ͬʱѪС��ɷֽ���鰷��Ѫ���ؼ����ģ�ʹѪ����Ƥϸ��������ϸ����϶�ӿ��������ʵ�����������Ҳ��������
��ˮ�����γɡ�
��ʼ��ˮ��Һ�������ڷ�С������Χ�ķμ�����֯���Ժ������࣬����������ϸ֧���ܣ��������������ݣ�����ͨ��/Ѫ����ע����ʧ�����γɵ���Ѫ֢��
2.����
Ѫ˨�γ� Solliday����Ϊ���������أ����п���ʹ���ڶ���Ӱ������ߣ���ʱ�����ƹ�����Ҳ������ҽԴ�Զ���Ӱ����ӡ�����Ӱ������������շ�ѪС���������γ�Ѫ˨���������������࣬���������ε�С�����������ѭ���ϰ���������ѪС�廹���ͷų�Ѫ���غ��鰷������֧���ܾ��Σ�Ӱ��ε�ͨ�����ܡ���
Ѫ˨�γ�����ά����ԭת��Ϊ��ά����ʱ��Ҳ���ͳ�Ѫ�ܻ����ģ����߿ɸ����ؾֲ�Ѫ�ܼ�֧���ܾ��Σ��ζ���ѹ���ߣ�����ëϸѪ��ͨ�����ӣ��Ӷ��������ݼ����ʳ�Ѫ��ˮ�ס���������ά���׳��������⣬˨����Ӱ���Ӫ��Ѫ�ܵ�Ѫ����ע���������֯�ṹ�ƻ������ʹ��˳Ӧ�Խ��ͣ��������������ۺ�������Malik����Ϊ��
Ѫ˨�γ�������Ѫ������Ѫͬʱ���ڣ��Żᷢ�����������ۺ�����
3.���ݱ�������������ɼ��� �����������ۺ�������ʱ������I�ͷ�����Ƥϸ���������ƻ����������ص�����ëϸѪ����Ѫ�����ϵ������ԣ����һ������ɢ�����Ƥϸ���ֻ������ƻ��˵�I����Ƥϸ��������ֱ��Ӱ���˷��ݱ���������ʵ����������������⣬���ڷ���ˮ��Һ���ӯ�����ݱ���������ʵĻ������н��ͣ����ݽ�����ή������˷����������½��������������ѡ�
���ˡ����������������Ժ����ĺ��������ۺ�����������һ��DZ���ڣ�����������ʵİ�˥����18��24h��������ʱ���Ͻ��ƣ���ˣ�������Ϊ���������ۺ����ķ����������ڱ���������ʲ����������¡�
4.ARDS�IJ����������ɶ�����֢ϸ��(����ϸ������������ϸ�����ܰ�ϸ����)�鵼�ķ���ֲ����Է�Ӧ����֢��Ӧʧ�����µķ�ëϸѪ��Ĥ���ˡ�����Ҫ��������Ϊ�ɷ�Ѫ��ͨ�����߶����µķ�������Һ�и��������ʵ�
��ˮ������Ĥ�γɣ��ɰ��зμ�����ά����
�ڳ����ٴ�֢״���ǰ18h�ڣ��δ�����ֲ�����������������ɢ�ڵij�Ѫ��
�β����������ݿ˺�18��72h�������أ�����Ϊ������Ҷ�ʳ�Ѫ�Բ��䡣���������طξ�����Ѫ��ɢ�ڵ�Ѫ˨˨���γɡ�����ˮ�ס�Ѫ�ܺ�֧������Χ��Ѫ�ͷ��ݳ�Ѫ��72h��ɼ���Ĥ��֧���ܷ��ף��̶�������ɢ����ά���Ըı䣬��������ֳ�Ըı��ͬʱ�����ڡ������ı���ص�ɹ������£�
(1)������(24��48h)�����ݺͼ���ˮ�ף�ëϸѪ�ܳ�Ѫ��I�ͷ���ϸ���ƻ���������Ĥ�γɡ�ˮ��Һ�ĵ��������ɷ���Ѫ������ơ���Ѫ�ܵ���Ƥϸ���������������ģ�δ��ϸ�����Ӵ�ȱ�ڡ������ڼ����ڿɷ��ֺ�ϸ������ʾ��Ѫ����Ƥ�������ݵ�©�졣����������Ƥϸ����ǿ����������ʹ��Ƥ����ݵ������Ա�������
(2)ϸ����ֳ��(3��7��)������ϸ������������ϸ������μ������Ĥ����������ϸ���������ں���Ϊ�������Ӧ����ʼѸ����ֳ�������������δ�ܵõ����ƶ����������������Դ̼��йء��ڴ��ڣ�����ϸ�������ڷ�Ѫ����Ƥϸ�����棬��Ѫ��
Ѫ˨�γ�����ʵ�ʵĸı����Ϊ��Ƥ�������������״�Ѫ�ܴ�Ϊ���ٻ��ܼ�ѹ�����ݡ����ڵļ����״�������ˮ���Լ�ϸ����ֳ��
(3)��ά��ֳ��(��7��10��)����Ĥ�ͷ��ݸ�����ά�������ݹ���ά����
(4)�������������ݳ����ţ����ݿ����������ݱ���ëϸѪ�ܸ������������֢��ͷ��ݼ�ëϸѪ����Ƥϸ�����ͣ��ڷ���ëϸѪ���ڿɼ�����ά���ס�ѪС�塢��ϸ���Ͱ�ϸ���������ַ���֯�У���ϸ���������ӣ��ں�ϸ���ͷ��ݱ��棬�ɼ�����ά�������ʸ��š�����ϸ���ĺ�Ⱦɫ�ʱ�֣����ܿ�϶���������������һ���ʧ�����������ţ������ṹ�ƻ������ſ��������Ԣ���ϸ���İ����г��ִ�С���ȵĿ��ݡ��ڷ���ǻ�ڣ��ɼ��������ŵİ�ϸ������ϸ��������ϸ��������Ģ���ϸ�����еķ���ǻ�л�����ˮ��Һ�������Һ��������μ��ʲ�����֯��϶�������ɳ��ֲ�ͬ�̶�ˮ�ף�ż��������ά�ͽ�ԭ��ά����ϡ������ҡ��������ش����ݽṹģ���������壬������Ƥϸ���ı���ɱ��ݺύ������ά�������ǡ�
����
���ƣ�ARDS�����ư����������ƻ����������Ⱦ�ʹ��˵ȣ��Լ���Ч����������֧�֣������Ǻ�������֧�֡��ۺ����ƴ�ʩ�Ͳ������£�����Բ�ͬ��������Ӧ���ƣ��ڼ�ʱ���ƣ�������л�еͨ�����ƣ��Ծ���Σ�������ĵ���Ѫ֢����ʱ���Ѫ���ͺ�������ѧ�仯���۾�����Һʱ�ϸ���Ʋ�Һ������ʱ���������Ѫѹ��Ѫ������ѧ�仯���ܸ���Ӫ�������ں������ƿ�ʼ72h�ڽ��У��ݼ�������ȫ������������״̬����ʱ���ֺʹ��������������ϰ������������ƣ��Ը�Ⱦ��Ӧǿ�����ݲ�ԭѧ��ϼ�ҩ����������ȷѡ��ҩ�
1.���� ����ARDS����Ҫ��ʩ�Ǽ�ʱ��������Ѫ֢����������ʱPaO2��8.0kPa(50mmHg)��Ӧ�����ơ�Ϊ�˷����ڷ�����ͨ��/Ѫ������ʧ��������Ӧ�����Ũ����(FiO2 0.6)�����ñǵ��ܻ����������������Ч������Ӧ���������֡����ڸ�Ũ�����������ɷ���֯����(���ж�)�����Ӧ�ڱ���PaO2��8.0kPa(60mmHg)��ǰ���£���������������Ũ�ȣ��������ط��ڷ���ʱ����ʹ���FiO2�����Ծ���ȱ��ʱ��Ӧ��ʱ���л�еͨ�����ơ�
2.��еͨ������ ��еͨ�����Ƶ�Ŀ���Ǿ���ȱ����ά���ʵ�����֯�������ȹ������ڣ��Դ�����֯������ͬʱ�����������ƣ�����µ�PaCO2����(���������ж�)����ͨ�����ֱ����ܲ�ܻ������п����Ӻ������������ƣ����������ڲ��÷���Ϯ��������ѹͨ�����ƣ�ȡ�ýϺ�Ч�����������߲����һ����չ�����輰ʱ�������ܲ�����ơ�
����Ļ�еͨ�����ƣ�ARDS���ø���/�����Ͷ���ͨ���ͼ�Ъָ��ͨ�����Ʒ�ʽ����ͳ�۵���ΪARDS���߷�ˮ�ף�����ͷβ��ŵȲ���ʹ㷺���ȷֲ������Ϊ��������Ѫ֢�����ʵ����������Ũ���⣬���л�еͨ������ʱ��Ӧ�����ýϴ�ͨ�����������ƵIJ��ԡ���������Ъ��ѹͨ��(IPPV)����ʱ�����ӳ�����(VT)��ʹή�ݷ��ݵ������ţ���ͨ�����ӷ�����ѹ��ʹ��ˮ��Ѹ�����ˡ���ˣ���еͨ������ʱ��������(VT)��������10��15ml/kg������Ƶ��Ϊ10��15��/min����������Ϊ1��1��1��1.5������Ũ��(FiO2)Ϊ0.6����ͨ������IPPV���ƣ�δ��ά�ֶ���Ѫ����ѹ��������Χ�����䵱FiO2��60%����PaO2����ά����60mmHg(8.0kPa)�����ϣ�����Ƭ��Ƭ�ν�����Ӧ�����к���ĩ��ѹ(PEEP)ͨ�����ƣ���ΪPEEP�����ӹ��ܲ����������η���ή�������ٷ��ڷ�����������ڽϵ͵�������Ũ��(FiO2)�������¾���ȱ����ͨ�����ź�����ĩ��ѹ(PEEP)5��10cmH20(0.49��0.98kPa)����������²�Ӧ����15cmH2O(1.47kPa)��ϣ���ܹ���FiO2��0.6�������£�����PaO260��70mmHg��SaO2��90%�����ƹ�����Ӧע�����PEEP���Ա�ﵽ����������ö���������С�����巽���ǹ۲쳱��˳Ӧ��(VT/ƽ̨ѹ-PEEP)�Ե���PEEP����VT���������£�������PEEPֵ�����ٴ�����ȶ�������״̬����(FiO2 0.4��PaO2��9.3kPa)��������(PEEP)ˮƽ���Ƚ���PEEPֵ0.5kPa(5cmH2O)����PaO2ˮƽ�ȶ����ԭˮƽ���ͣ�20%���ɸ��ݲ������롣
������������ͨ��ģʽ���Խϴ�ͨ�������л�еͨ�����ƵIJ��ԣ���Ȼ��һ���̶��Ͽ��Դﵽ��߶���Ѫ����ѹ��Ŀ�ģ������ٴ�ʵ����ֻ��������ƹ��������������ͷ�����(�μ������ף����أ��ݸ����ף�Ƥ������������)���Լ�ѭ���ϰ�(���������ͺ͵�Ѫѹ)����������ͷ������ͣ��Ӿ���֯ȱ�����о�����ARDS���߷β�������ʲ����ȷֲ������ˮ�ͷ�ʵ��Ϊ�����ȵİ�Ƭ״�ֲ��������ڼ�������ά������Ҫλ�������´���λ������λ���λ�ڱ������β�����Чͨ��������ܽ�ռȫ�ε�20%��30%������Ը߳��������л�еͨ�����ƣ���Ȼ��������������·�����(�����ͷ�����)������ʵ����ָ߳�������������ѹ��ͨ�����ƶ������λ����֯���������Ӿ缱�Է����ˣ����������ͷ�ˮ�ס�
Ŀǰ�ཨ����õͳ�����ѹ��֧��ͨ��(pressure support ventilation��PSV)ģʽ�ͻ�еͨ�����ƣ�������(VT)4��7ml/kg������Ƶ�ʣ�30��/min������ƽ̨ѹ��30��35cmH
2O(0.29��0.34kPa)���ʵ��ӳ�����ʱ�䣬��ϵ���������Ũ��(FiO
2)�Ծ������ص���Ѫ֢�����ô���ͨ�����Ʒ������������Ի������߳�����������ѹ����������ͷ����ˣ��������ڷ����˵����Ϻ���ѭ���ϰ�������֯ȱ�������Dz��õͳ�����ѹ��֧��ͨ���ĵ�ͨ������еͨ�����ƻ����
������̼���ų����ޣ����²�ͬ�̶ȵĸ�̼��Ѫ֢������Ŀǰ��Ϊ���ARDS������������Ҫԭ�������ص���Ѫ֢�����ʵ���������Ѫ֢��ǰ���£���ʹ����һ���̶ȵĸ�̼��Ѫ֢���Ի��岻���������������ARDS������ת��������ȱ���õ�������ͬʱ��������
������̼������ų�����ˣ������еͨ������ʱ���������Ը�̼��Ѫ֢(permissive hypercapnic acidemia��
PHA)�IJ��Ը��������Ѫ
������̼��ѹ(PaCO
2)ֵ������Ӧ��һ����(100��120mmHg��13.3��16kPa)����PaCO
2���ߵ��ٶ�Ӧ��10mmHg/h���Ա�ͨ�����������ʹPHֵ�������Խ��͡���ѹ���ߵĻ��߲��˲��ñ�ģʽ����ͨ�����ƣ�ȱѪ�����ಡ�����������ҹ����ϰ�����Ӧ���á�
���������õ�������ѹ��ͨ��ģʽ�а������ٷ���ͨ��(inversive ratio ventilation��IRV)���ں���Ƶ���ȶ������£���������Ƶ��Ϊ��/����1.5��1���������ʱ����Խϳ�������ʱ����Խ϶̣�IRV��ѹ������(��ѹ��)ģʽ����ͨ�����ơ��÷������ŵ���ʹ���ڲ�ͬ���䲿λ������ѹ���ֺ㶨��������ѹ�������ˣ��Ҵ�����֯��ԭ����˳Ӧ�Ըı���Զ����ӳ������������ڻ����̼��Ѫ֢�������ܻ�������Դ�Ժ���ĩ��ѹ(auto-PEEP)���Ҳ��˲�����ϣ���Ҫ������Ӧ�ǽ��;��������������ŷ�����ͨ������ѹ��֧�ֻ�ѹ������ͨ�������������ݷ�����Ϣ��λ�ò����Զ�������������ѹ���ͷ�ͨ��(airway pressure release ventilation��APRV)��˫ˮƽ������ѹͨ��(BiPAP)��Ƶͨ��(HFV)������������������ȱ������ֵ������������֤������Խ�ԡ�
��еͨ�����ƹ�����Ӧע�������ѭ������˽ⲡ���ݱ䣬��ʱ�������������Ʋ��������ֺ������Ͳ��˺�������ͬ��(�˻��Կ�)ʱ��Ӧ����ԭ���ɲ��������磺������(����)5��10mg��ע��ע��������������(
Midazolam)��
�������(Disopinfol)����Ҫʱ�ü����ɳڼ�����
�����(Vecuionium)��
3.�ǻ�еͨ������任����
(1)����Ĥ������(extracorporeal membrane oxygenation��EMO)������Ĥ�������������Ѷ��꣬����Ч���������ڳ���Ļ�еͨ�����ƣ�����ŵ���ʹ�ã���ż���ڲ������ط������߽��л�еͨ�����Ƶĸ������ơ�
(2)������������(intravenous oxygenator��IVOX)�����������п���ά���Թɾ���������ǻ��������˽������彻���������彻����̫�٣��Ҿ������������IJ��뽵��������ָ�����������������������Ⱦ��˨���Ȳ���֢��
(3)����Һ��ͨ��(partial liquid ventilation��PLV)����̼��Һ(perfluorocarbon)�������õ���������Լ���Ե͵ı����������������彻��ý�����Һ��ͨ�����ƣ�����Һ��ͨ��������Լ�൱�ڹ��ܲ������ķ�̼��Һ(Լ30ml/kg)ע����ڣ���ϳ����еͨ�����ƣ��������������������ۺ���������ȡ��һ���ɹ���
(4)������λ(prone position)��ARDS����ȡ������λ���л�еͨ�����ƣ��ɽ�һ����������״̬����Ȼҽ�ƻ���������һ�����ѣ��������ս��ձ顣ARDS���߸�����λʱ�α��ν���ͨ��/����ʧ�������ڷ����������Ϊ���أ�ȡ����λʱ���ò�λ�ľ���ѹ������������ѹ�������ڷ��ھ���ͨ����Ѫ���ֲ�������������λ��������˸��Ƶ���Ѫ֢��
4.һ��֧������
(1)Һ���Ѫ������ѧ������Һ���������Ҫ���ڣ�����Һ����ƽ�⣬���ӷ�ëϸѪ��ѹʹҺ����©�������ز��鲢Ӱ��Ԥ��Ӧά�ֽϵ͵ķ�ѭ��ѹ������ȷ���ղ�Һ��[20��25ml/(kg•d)]���Խϵ���ЧѪ������ά��ѭ�����ܡ������ˮ�Ӿ硣���˲��þ�����Һ����1.3%��1.5%Nacl��Һ�������˲��ý�����Һ�����Ե���ѹ�߸��ڽ�����Һ����
������
Ѫ��������ƶѪ(Ѫ�쵰�ף�100g/L)�ߣ�Ӧ���ϸ��������ѪҺ������һ����ѭ��Ѫ�������Ӱ���еͨ�����ƺΡ������ܣ����Ӧ����Ѫ������ѧ��⣬����ͨ��Swan-Gang�ζ������ܼ��ζ���Шѹ[ά����14��16cmH
2O(1.37��1.57kPa)]�����������Լ���Ҫʱ����ʳ�ܳ����Ķ�ͼ��ʳ�ܶ�����(Dopler)ɨ�裬��������ӯ���������������Ϊ�ٴ�Һ�����ָ��IJο���
(2)ȫ��Ӫ��֧�ֺ���������֧�֣����ߴ��ڸߴ�л״̬��Ӫ�����������º�����ƣ�ͺͶ���������˥�ߣ���Ժ�ڸ�Ⱦ�����������ߣ���Ӧ��ʱ����ȫ��֧�����ơ������ݲ��龡��ָ�θ��Ӫ��;��������������θ�����ܵĻָ�����ȡӪ�����ʣ���ʳ��Ӧ��֬���ᣬ���ʺ����ᣬ
��������
�Ȱ�����ά���صȳɷ֡�����ȷ������������θ����Ӫ��;���ౣ���������Ĥ���Ϲ��ܡ�����θ������Ⱥת�ƽ�������ϵͳ������Ժ�ڸ�Ⱦ�ķ������ᡣ
5.ҩ������
(1)��Ƥ�ʼ��أ���Ƥ�ʼ��ؾ��й㷺�Ŀ���֢�ͼ���ëϸѪ��Ĥͨ�����á���Ӧ��������ARDS���������ٴ�������Ϊû�ܸ��ƻ���Ԥ�����������Ӹ�Ⱦ�Ȳ���֢�Ŀ��ܣ�����ٴ�Ӧ���������顣Ŀǰ������Ϊ��Ƥ�ʼ������ƿ��������ڼ��Ի����������ף�θ���������룬���ж��������������ˣ�ŧ�����ݿ˺�֬��˨�������ARDS��������Ϊ��Ƥ�ʼ���������Ч�ı�������ڸ������������ƵĹ۲��������ǵ������ڲ����п�������֢���ʵij����ͷţ�����ͣ����Ƥ�ʼ��أ����ܻᵼ�²��������п�����Ҫ�ӳ���Ƥ�ʼ��ص�����ʱ�䡣��ʼ�ü���������(methylprednisolone)5mg/kg��ÿ6Сʱ1�Σ����൱������������Ƥ�ʼ������ơ����������������ڲ�����ڣ����羭����֧������1�ܺ���˥������ת��������ARDS�Ĵٷ������ѵõ����ƣ�����Ⱦ���ٴ������ߣ����ü���������2mg/(kg•d)������1�������ң���Ϊ�ɸ��Ʋ��飬���������ʡ�
�����������Կ�����
ǰ������E1(ProstaglandinE��PGEl)�������(Buprofen)����ͪ�ɿɼ�(Pentoxifylin)����ά���ӵ���(Fibroneefin��FN)��
����(
Heparine)��Ч���ޡ�һ������(NO)��Prostacyclin��
�廷������(Ambroxol)�Ⱦ߿����������ã���������ARDS�д��۲졣
(2)�α�����Լ�(pulmonary surfactant��PS)����ȡ��Ȼ��ȡ������ķα�����Լ����û����油���ƣ����Ը��Ʒ�˳Ӧ�ԣ�����һ�����ߵ������ã�������ֲ����ƶ�����PSȱ����������������������ۺ�������һЩ�����ɹ��ı�������ARDS�IJ����Լ��������ƱȽϸ��ӡ�������PS���Ƶ�ȷ��Ч���͵�λ���ڽ����о���
(3)�����Կ�������������ȼ����ķ�չ��Ϊ����
ADRS����µ�;����ϣ��������ٿ��ڶ���(Antiendotoxin)-���鿹�ڶ��ص���HALA��Ӧ���ڸ������Ը˾���Ⱦ��Ѫ֢���ڶ��������
ADBS���ڿ�ϸ������(anticytokine)��������Ѫ֢���ݿ˻�������¡����TNF-�����壬�������и��ƣ�����IL-12�Ģ����ٴ��۲�����м�����ص������ʸ��ƣ��ۿ�����(anticomplement)�����ٴ��۲���δ�ܽ��Ͳ�����ԣ�ʹ�ð�ȫ����Ч�����Ǹ��������Կ���������Ȼ�ڶ���ȡ��һ������Ч����������ٴ��۲���δ��֤ʵ����Ч��������Ч�ı�������ȱ���й�ģ����������δ�ܱ������о������ظ�������������½��ۡ���������Ҫ��ARDS��չ�����и�����֢���������������õ�ʱ������е�һϵ�л������������˽⼰��Ƹ������Ķ������ٴ��۲췽����������Ч����